

Ги запознаваме ретките болести во охридско – струшкиот регион: Акрокалозален синдром и Амелогенезис имперфекта
Во рамките на кампањата за запознавање на ретките болести, ве запознаваме со две дијагнози. Акрокалозалниот синдром, е ретко генетско нарушување кое е очигледно уште при раѓањето. многу засегнати лица имаат малформации на черепот и лицето или карактеристични абнормалности на прстите на рацете и нозете. Амелогенезис имперфекта е ретко, наследно нарушување кое се карактеризира со абнормално формирање на емајлот (површниот слој на забот) и во млечна и во трајна дентиција.
Во месецот со најмалку дена во годината, во месецот кој оваа година е редок по своите ретки 29 дена и во месецот кој пациентите со ретки болести го идентификуваат како СВОЈ, по шести пат ќе се обидеме поблиску да ве запознаеме со ретките болести од кои заболува и со кои се соочува и живее населението од охридско – струшкиот регион.
Да ги дознаеме предизвиците и проблемите со кои се соочуваат.
Како ја добиваат дијагнозата?
Дали има некаков лек?
Колку и дали воопшто се пристапни и достапни лековите?…
АКРОКАЛОЗАЛЕН СИНДРОМ/ ACROCALLOSAL SYNDROME
MKБ-10 Q04.0
ПРЕВАЛЕНЦА
Преваленцата на aкрокалозалниот синдром е приближно 1 / 1 000 000 новороденчиња.
ВОВЕД
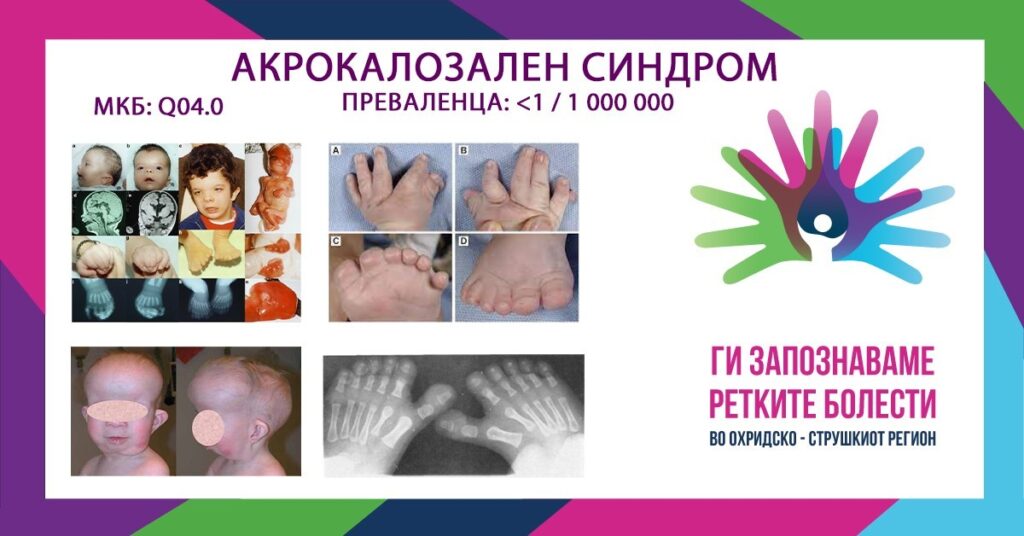
Акрокалозалниот синдром е ретко генетско нарушување кое е очигледно уште при раѓањето. Придружните симптоми и наоди можат да бидат променливи, вклучително и меѓу засегнатите членови од исто семејство. Сепак, нарушувањето типично се карактеризира со неразвиеност (хипоплазија) или отсуство (агенеза) на густата лента на нервните влакна што ги спојува двете хемисфери на мозокот (corpuscallosum) и умерена до тешка ментална ретардација. Покрај тоа, многу засегнати лица имаат малформации на черепот и лицето или карактеристични абнормалности на прстите на рацете и нозете. Карактеристичните краниофацијални абнормалности можат да вклучуваат невообичаено голема глава со истакнато чело, широко распоредени очи, спуштени набори на очните капаци, мал нос со широк назален мост и малформирани уши. Повеќето засегнати поединци, исто така, имаат карактеристични дигитални малформации, како што е присуството на дополнителни прсти на рацете и нозете (полидактилија) и спојување. Можат да бидат присутни и дополнителни физички абнормалности, вклучувајќи нарушување на растот што резултира со низок раст. Иако е автосомно-рецесивно наследување, акрокалозалниот синдром често се чини дека се јавува случајно од непознати причини. Акрокалозалниот синдром ги погодува и машките и женските деца во релативно еднаков број. Нарушувањето првично било пријавено во 1979 година (А. Шинзел). Во медицинската литература се евидентирани над 25 случаи.
ЕТИОЛОГИЈА
Мутациите во кинезин KIF7 (15q26.1) и во транскрипцискиот активатор GLI3 (7p14.1) се одговорни за акрокалозалниот синдром. ACS е автосомно-рецесивно заболување и затоа постои 25 % ризик од повторување за следната бременост. Ако генската мутација во гените на кинезин/транскрипцискиот активатор е идентификувана кај засегнат брат или сестра, молекуларната генетска дијагноза може да се направи по земање примерок од хорионските ресички.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Кај индивидуи со акрокалозален синдром, опсегот и сериозноста на поврзаните наоди можат да бидат исклучително променливи. Сепак, во сите пријавени случаи до денес, нарушувањето се карактеризира со неразвиеност (хипоплазија) или отсуство (агенеза) на густата лента на нервните влакна што ги спојува двете хемисфери на мозокот (corpuscallosum), како и умерена до тешка ментална ретардација. Во некои случаи, малформациите на мозокот можат да бидат поврзани со дополнителни компликации, како што се ненадејни епизоди на неконтролирана електрична активност во мозокот или хидроцефалус, состојба во која нарушениот проток или апсорпција на течноста што циркулира низ шуплините на мозокот и ’рбетниот канал (цереброспинална течност CSF), потенцијално доведува до зголемување на притисокот во мозокот. Поединците со акрокалозален синдром, исто така, можат да имаат абнормално намален мускулен тонус (хипотонија) и да доживеат тешко психомоторно нарушување или забележително доцнење во развојот на одредени физички, ментални или бихејвиорални вештини кои обично се стекнуваат во одредени фази. Над 50 % од засегнатите поединци исто така имаат абнормални доцнења во растот, што често резултира со низок раст. Поединци со акрокалозален синдром, исто така, можат да имаат абнормалности на очите. Тие можат да вклучуваат широко распоредени очи (хипертелоризам), надолни коси набори на очните капаци, вертикални кожни набори кои можат да ги покријат внатрешните агли на очите и овенати горни очни капаци (птоза). Во некои случаи, можат да бидат присутни дополнителни очни дефекти, како што се внатрешно отстапување на едното око кон другото (конвергентен страбизам или езотропија), абнормалности на обоените слоеви на мрежницата и дегенерација на нервите кои пренесуваат импулси од мрежницата до мозокот (оптичка атрофија).
Акрокалозалниот синдром исто така, обично се карактеризира со карактеристични малформации на прстите на рацете и нозете. На пример, кај многу засегнати индивидуи, може да има дуплирање или присуство на дополнителни палци. Дополнителните абнормалности на прстите често вклучуваат плетење или спојување (синдактилија) на одредени прсти, особено од првиот до третиот прст, и неразвиеност или спојување на зафатените нокти. Некои засегнати доенчиња можат да имаат структурни срцеви малформации при раѓање (вродени срцеви маани). Тие обично вклучуваат абнормален отвор помеѓу преградите на преткоморите, односно коморите, како и нарушување на срцевите залистоци. Дополнително, кај некои засегнати поединци, може да се развие кила во препонската регија (ингвинална хернија) или во предниот абдоминален ѕид околу папокот. Некои засегнати мажи можат да имаат и генитални абнормалности, како што се неспуштени тестиси, абнормално поставување на уринарниот отвор или невообичаено мал пенис. Кај некои доенчиња со акрокалозален синдром, периодот непосредно по раѓањето може да биде комплициран со епизоди на напади, тешкотии во хранењето и зголемен ризик од респираторни инфекции. Во некои случаи, респираторните инфекции, како и тешкотиите со дишењето (отежнато дишење), недостатокот на доволно снабдување со кислород до ткивата и други поврзани абнормалности можат да доведат до потенцијално опасни компликации.
Со оглед на високата варијабилност на фенотиповите, кои било два од следниве критериуми ја поттикнуваат дијагнозата на ACS:
- целосно или делумно отсуство на corpuscallosum
- мали краниофацијални аномалии
- умерено до тешко доцнење во развојот со хипотонија
- полидактилија
Молекуларното генетско тестирање може да се користи за да се потврди дијагнозата и да се води генетско советување. Во некои случаи, како на пример, во семејства со членови кои претходно биле дијагностицирани со нарушување, лекарот може да се посомнева на акрокалозален синдром пред раѓањето (пренатално) врз основа на одредени специјализирани тестови, како што се ултразвук или фетоскопија. Антенаталната дијагноза се заснова на ултрасонографија од 20-та недела од бременоста и магнетна резонанца (МРИ) на фетусот. Дијагнозата обично се поставува или потврдува при раѓањето врз основа на темелно клиничко испитување, идентификација на карактеристичните физички наоди и разниспецијализирани тестови. Таквото тестирање може да вклучува рендгенски студии, компјутерска томографија или магнетна резонанца, или други студии за да помогнат во откривање или карактеризирање на одредени малформации кои можат да бидат поврзани со нарушувањето (агенеза или хипоплазија на корпус калозум, одредени краниофацијални абнормалности, полидактилија и синдактилија). Може да се препорача и темелна срцева евалуација за да се откријат какви било срцеви абнормалности кои можат да бидат поврзани со нарушувањето: електрокардиографија, ехокардиографија, срцева катетеризација. Диференцијалната дијагноза вклучува: Грејг-цефалополисиндактилиски синдром, синдром орофациодигитален тип I, синдром Смит–Лемли–Опиц, синдром Рубинштајн–Тајби, синдром Горлин, Аикарди синдром, како и синдром Мекел–Грубер и хидролеталус во екстремни форми.
ПОСЛЕДИЦИ
Прогнозата зависи од тежината на малформациите, хипотонијата и од појавата на напади.
ТРЕТМАН
Третманот на акрокалозалниот синдром, тип Шинзел, е насочен кон специфичните симптоми кои се очигледни кај секој поединец. При таквиот третман можат да бидат потребни координирани напори на тим од медицински професионалци, како што се педијатри, хирурзи, лекари кои дијагностицираат и лекуваат невролошки нарушувања, лекари кои се специјализирани за срцеви заболувања, лекари кои дијагностицираат и третираат нарушувања на скелетот, зглобовите, мускулите и сродните ткива, офталмолози и/или други здравствени работници. Специфичните терапии за овие лица се симптоматски и поддржувачки. За оние со хидроцефалус, при раната операција може да се вметне цевка (шант) за да се дренира вишокот цереброспинална течност (CSF) од мозокот и во друг дел од телото каде што може да се апсорбира CSF. Во некои случаи, може да се препорача и хируршка интервенција за да се поправат одредени краниофацијални малформации, полидактилија и синдактилија и/или други физички абнормалности потенцијално поврзани со нарушувањето. Дополнително, за оние со вродени срцеви маани, може да биде неопходен третман со одредени лекови, хируршка интервенција и/или други мерки. Изведените хируршки процедури ќе зависат од сериозноста и локацијата на анатомските абнормалности, нивните придружни симптоми и други фактори. Лекарите можат редовно да ги следат засегнатите доенчиња и деца за да помогнат да се обезбеди брзо откривање и ран агресивен третман на респираторните инфекции. Дополнително, во некои случаи, можат да се препорачаат одредени превентивни мерки за оние кои можат да бидат склони кон повторени респираторни инфекции. За засегнатите доенчиња кои развиваат респираторен дистрес, третманот може да вклучува различни мерки за поддршка, вклучително и соодветна терапија со кислород. Управувањето со болеста може да вклучи и супортивни терапии за да помогне да се обезбеди правилен внес на калории и исхрана кај оние со тешкотии во хранењето. Во некои случаи, може да се препорача третман со одредени антиконвулзивни лекови за да се спречат, намалат или контролираат нападите. Раната интервенција може да биде важна за да се даде сигурност дека децата со нарушување ќе го достигнат својот потенцијал. Специјалните услуги кои можат да бидат корисни вклучуваат специјално образование, физикална терапија и/или други медицински, социјални или стручни услуги. Генетското советување ќе биде од корист за засегнатите поединци и нивните семејства.
АМЕЛОГЕНЕЗИС ИМПЕРФЕКТА/ AMELOGENESIS IMPERFECTA
MKБ-10 K00.5
ПРЕВАЛЕНЦА
Амелогенезис имперфекта е релативно ретка аномалија. Податоците од литературата покажуваат 43 : 10 000 во Турција, 14 : 10 000 во Шведска, 10 : 10 000 во Аргентина, 1,25 : 10 000 во Израел. Средната светска вредност на преваленцата изнесува < 0,5 % (< 1 на 200). Хипопластичниот тип на амелогенезис имперфекта се јавува во 60 – 73 % од случаите, хипоматурациониот тип кај 20 – 40 %, а хипокалцификацискиот тип во 7 % од случаите.
ВОВЕД
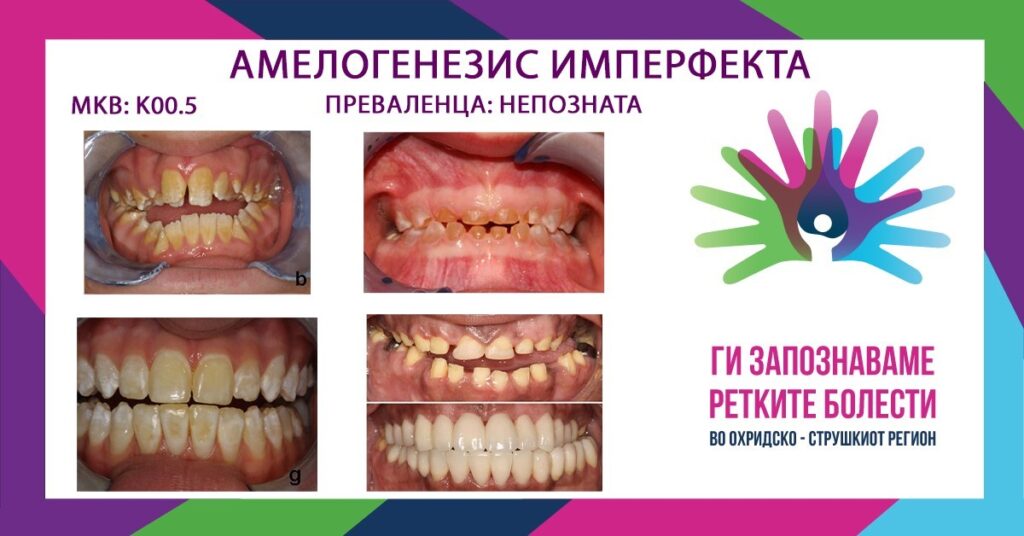
Амелогенезис имперфекта претставува ретко, наследно нарушување кое се карактеризира со абнормално формирање на емајлот (површниот слој на забот) и во млечна и во трајна дентиција. Клиничката слика е разнолика, од тенок и мазен емајл до груб емајл со вдлабнатини во облик на јамички или жлебови со жолто-кремава боја. Терминот „амелогенезис имперфекта“ се однесува на дефекти на емајлот во отсуство на други манифестации, но постојат случаи во кои претставува еден од симптомите во склоп на некоја системска болест или синдром. Многу пореметувања на метаболизмот, како и генетски состојби имаат влијание на изгледот на забите. Абнормалноста во структурата на забите може да биде поврзана со општи пореметувања во калцификацијата (хипотиреоидизам, хиперпаратиреоидизам), тешки болести во детска возраст (хронични бубрежни заболувања, гастроинтестинални пореметувања), инфекции (рубеола), штетни делувања од лекови (тетрациклин), интоксикација (флуороза на заби), кожни промени (ектодермална дисплазија), пореметувања во синтезата на коскениот матрикс (остеогенезис имперфекта), хормонски пореметувања (хипопитуитаризам) и др.
ЕТИОЛОГИЈА
Забниот емајл се состои од високо организирани хидроксиапатитни кристали кои сочинуваат 87 % од волуменот на емајлот. Кристалите се протегаат од дентинот кон површината на забот, а организирани се во призми. Тоа се ацелуларни творби без можност за внатрешна репарација. Емајлот го создаваат амелобластите при процес на амелогенеза, а се дефинираат од внатрешниот емајлов епител. Процесот амелогенеза е под генетска контрола. Одредени мутации на генот се одговорни за развој на аномалии на емајлот. Аномалиите се наследуваат автосомно-доминантно, автосомно-рецесивно или Х-врзано наследување.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Класификацијата на амелогенезис имперфекта е претставена преку четири основни типа на аномалија со поттипови:
- ТИП 1: хипопластичен амелогенезис имперфекта (хипоплазија на емајл) – најзастапен тип, кој настанува поради пореметувања во секреторната фаза на амелогенезата. Се карактеризира со помала количина на емајл, нормална минерализација и матурација. Бојата зависи од степенот на редукција на емајлот. Ако дебелината е незначителна, бојата е нормална, а ако дебелината на емајлот е значително редуцирана, забот има жолтеникаво-кремава боја. Емајлот може да биде груб, мазен, вдлабнат, локално или генерализирано хипопластичен. Доколку зафатеноста е генерализирана, коронките се мали и имаат коничен или цилиндричен облик, а измеѓу некои од забите нема контакт. Основен клинички проблем е што емајлот е многу тенок и брзо се губи, а дентинот е изложен на абразија. Кај 60 % од пациентите се јавува и отворен загриз.
- ТИП 2: хипоматурациски амелогенезис имперфекта (хипоматурација на емајл) – настанува поради пореметување во фаза на минерализација на емајлот. Кај млечните заби, емајлот е кредесто-бел, а кај трајните варира од кредесто-бело до жолто-кремава боја. По никнувањето, брзо доаѓа до отпаѓање на слабо минерализираниот емајл, особено во инцизалниот дел.
- ТИП 3: хипоминерализациски амелогенезис имперфекта (хипокалцификација на емајл) – настанува поради пореметување во фазата на минерализација на емајлот. Емајловиот органски матрикс е нормално формиран, но несоодветно минерализиран што резултира со квалитативни дефекти. Во моментот на никнување, забот има емајл со нормална дебелина, бела или жолто-кремава боја, но слабата минерализација го прави емајлот мек и подложен на абразија. Кај 60 % од пациентите се забележува и отворен загриз.
- ТИП 4: хипоматурациско-хипопластичен амелогенезис имперфекта со тауродонтизам – претставува комбинација од хипоматурација и хипоплазија на емајл, а настанува поради пореметување на склеротични и матурациски фази на амелогенеза. Тауродонтизмот се појавува на моларите, а на останатите заби има волуменозни пулпини комори.
ПОСЛЕДИЦИ
Редовните прегледи се клучни за прилагодување на третманот кој може да варира и зависи од тежината на состојбата и потребите на пациентот. Раната дијагноза и соодветниот третман можат значително да го подобрат квалитетот на живот на лицата со оваа аномалија.
ТРЕТМАН
Потребен е мултидисциплинарен тераписки пристап при лекување на лица со амелогенезис имперфекта – тим од специјалисти по детска и превентивна стоматологија, ортодонција, протетика, а по потреба и максилофацијална хирургија. Превенцијата е најважна особено кај пациенти со нарушен интегритет на тврдите забни структури. Целта на педодонтот е да му влее позитивен став на пациентот кон оралното здравје и постепено да му го образложи сложениот третман по завршување на растот. Превенцијата и редовните прегледи се неопходни бидејќи зголемен е ризикот за појава на кариес и во млечна и во трајна дентиција. Одржувањето на соодветна орална хигиена кај пациентите со амелогенезис имперфекта е отежнато поради преосетливоста. Се препорачува примена на топла вода при четкање на забите и користење пасти со флуориди, што би ја редуцирало преосетливоста и појавата на кариес. Ортодонтот има клучна улога да го прати развојот и да ги постави забите во идеална позиција. Уште од најмала возраст со примена на превентивни и интерцептивни помагала, миофункционални вежби, потоа со мобилни, функционални или фиксни апарати, а кај адултни пациенти, ако е потребно, се применува и ортодонтска терапија со ортогнатна хирургија. По ортодонтскиот третман особено важен е постортодонтскиот период со редовни контроли за да не дојде до рецидиви. Протетската терапија кај пациентите со амелогенезис имперфекта претставува конечна санација, естетска реставрација и трајно решение на аномалијата. Потоа следи фаза на правилно одржување орална хигиена, одржување здрав пародонт и стабилна оклузија.