

Ги запознаваме ретките болести во охридско – струшкиот регион: Буфталмус и Дишен мускулна дистрофија
Здружението „Ретко е да си редок“ секој ден во февруари ќе објавува текстови со кои јавноста поблиску ќе се запознае со ретките болести од кои заболуваат и со кои се соочуваат луѓето во нашата држава, годинава конкретно, населението од охридско – струшкиот регион.
Во месецот со најмалку дена во годината, во месецот кој оваа година е редок по своите ретки 29 дена и во месецот кој пациентите со ретки болести го идентификуваат како свој, по шести пат Здружението „Ретко е да си редок“ секој ден во февруари ќе објавува текстови со кои јавноста поблиску ќе се запознае со ретките болести од кои заболува и со кои се соочува и живее населението од охридско – струшкиот регион.
Да ги дознаеме предизвиците и проблемите со кои се соочуваат. Како ја добиваат дијагнозата? Дали има некаков лек? Колку и дали воопшто се пристапни и достапни лековите?…
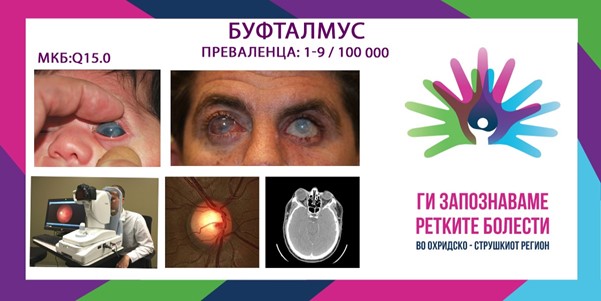
БУФТАЛМУС/ BUPHTHALMOS
MKБ-10 Q15.0
ПРЕВАЛЕНЦА
Се смета дека проценетата фреквенција е 1 – 9 случаи на 100 000 раѓања.
ВОВЕД
Зборот „буфталмус“ потекнува од грчкиот збор „волови очи“. Оваа состојба е поврзана со зголемен интраокуларен притисок (ИОП). Во моментов, терминот „буфталмус“ се користи за да се опише видливото проширување на очното јаболко откриено при раѓање или набрзо потоа, поради каков било неконтролиран глауком во раното детство.
ЕТИОЛОГИЈА
Буфталмусот најчесто се јавува поради примарен вроден глауком. Други состојби кои можат да предизвикаат покачен IOP во раното детство, исто така, можат да предизвикаат буфталмус. Ова ги вклучува Sturge-Weber синдромот, неврофиброматоза и аниридија.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Најчеста причина за буфталмус е примарен вроден глауком (почеток на раѓање) или примарен инфантилен глауком (почеток по раѓање до 3 години). Буфталмус обично не се забележува кај глауком со почеток по 3-годишна возраст. Класичните симптоми на вроден и инфантилен глауком вклучуваат кинење, фотофобија и раздразливост. Родителите можат да забележат замаглена рожница или зголемување на големината на рожницата. На преглед, лекарите забележуваат дека има зголемен дијаметар на рожницата, длабока предна комора и зголемување на големината на глобусот. Испитувањето на рожницата открива присуство на едем на рожницата, со руптури на Десеметовата мембрана, познати како Хаабови стрии. Интраокуларниот притисок е покачен и може да има чадови на оптичкиот диск.
ПОСЛЕДИЦИ
Зголемениот ИОП се манифестира како буфталмус кај педијатриските пациенти, така што компликациите се поврзани со висок ИОП. Овие компликации вклучуваат глаукоматозна оптичка невропатија, оштетување на рожницата, амблиопија и рефрактивна грешка. Раното намалување на ИОП може да ги спречи овие компликации, но доколку се присутни, амблиопијата и рефрактивната грешка ќе треба да се третираат одделно. Оштетувањето на рожницата може да биде неповратно и во иднина можно е пациентот да има потреба од операција. Ако пациентот има буфталмус поради вроден глауком кој останува недијагностициран на долг временски период, визуелната прогноза генерално е многу лоша. Губењето на видот поради вроден глауком е неповратно и сериозно. Раната хируршка интервенција за овие пациенти е поврзана со подобри визуелни исходи. Ако се третира рано, постои недостаток на глаукоматозна прогресија во повеќето случаи во првата година од третманот. Во текот на остатокот од животот на пациентот, речиси половина покажуваат понатамошна прогресија, нагласувајќи го фактот дека неопходно е да се врши редовен офталмолошки мониторинг во текот на животот на пациентот.
ТРЕТМАН
Главната цел на третманот е да се намали ИОП за да се спречи прогресивна заматеност на рожницата и глаукоматозна оптичка атрофија и со тоа да се зачува постоечкиот вид. Најдефинитивниот третман на буфталмосот е хируршки. Медицинскиот менаџмент е индициран за контрола на ИОП за чистење на рожницата за време на операцијата.

ДИШЕН МУСКУЛНА ДИСТРОФИЈА (ДМД)/ DUCHENE MUSCULAR DYSTROPHY (DMD)
МКБ-10 G71.0
ПРЕВАЛЕНЦА: 1 – 9 случаи на 100 000 лица
ИНЦИДЕНЦА: 1 на 3 500 до 5 000 машки новороденчиња
ВОВЕД
Дишен мускулна дистрофија (ДМД) е Х-врзана, рецесивна, генетска, невромускуларна болест. Таа се карактеризира со брзо прогресивно слабеење на мускулите и дегенерација на скелетните и срцевите мускули. Примарно се појавува кај машката популација, додека пак, женските лица се носители на болеста и најчесто се без симптоми. Но, кај мал процент од женските носители можат да се манифестираат слаби форми на болеста.
ДМД се јавува кај приближно 1 на 3 500 до 5 000 машки новороденчиња низ светот. Бројот на засегнати женски лица со ДМД е многу помала бидејќи тие типично се само носители и се без симптоми на болеста. Инциденцата и преваленцата на ДМД можат да варираат во зависност од популацијата бидејќи фреквенцијата на мутациите кои предизвикуваат ДМД, можат да се разликуваат од регион до регион. Тешко е да се одреди точната преваленца бидејќи болеста честопати е недоволно дијагностицирана, особено во помалку развиените земји.
ЕТИОЛОГИЈА
Дишен мускулна дистрофија е предизвикана од грешка (мутација, делеција) во генот DMD кој е лоциран на Х-хромозомот. Хромозомот Х е еден од двата полови хромозома, а машките имаат еден Х и еден Y хромозом, додека жените имаат два Х-хромозома. Генот DMD е одговорен за производство на протеин наречен дистрофин, кој помага да се одржи структурниот интегритет на мускулните влакна и ги штити од оштетување при контракција.
ДМД е Х-врзана, рецесивна болест, па наследувањето е на следниот начин:
Жена-носител има една нормална и една мутирана копија на генот DMD. Таа најчесто е без симптоми, но има 50 % шанса да го пренесе мутираниот ген на нејзините потомци
Маж кој има наследено мутиран ген од мајката ќе развие ДМД. Бидејќи има само еден Х-хромозом, тој нема нормална копија на генот DMD за да ја компензира мутацијата
Здрава жена има две нормални копии на генот DMD и нема симптоми на ДМД
Важно е да се забележи дека моделот на наследување на ДМД може да биде сложен, а лицата со семејна историја на ДМД треба да побараат генетско советување за да го разберат нивниот ризик од наследување или пренесување на болеста.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Првите симптоми и знаци на ДМД кај момчињата обично се појавуваат рано во детството и обично стануваат забележителни на возраст од 3 до 5 години. Во некои случаи, симптомите можат да бидат присутни од самото раѓање или да се појават подоцна во детството. Првите знаци често вклучуваат задоцнети развојни моторни достигнувања, како што се одење, трчање и/или скокање. Одењето често е паткасто и се карактеризира со широко, нестабилно одење и тешкотии во одржувањето на рамнотежа. Генерално, децата со ДМД никогаш не успеваат да трчаат или скокаат, а качувањето по скали станува потешко и детето почнува често да паѓа. Мускулната слабост обично започнува во долните екстремитети и карлицата, и се шири кон рацете, вратот и другите делови. Како што болеста напредува со текот на времето, мускулната слабост станува сѐ поизразена, што доведува до значителна функционална попреченост, губење на подвижноста и независноста, и потреба од помагала за одење, како што е инвалидска количка. Засегнатите лица исто така можат да имаат потешкотии и со изведување на фините моторни вештини, како што се закопчување на кошула, пишување, фаќање предмети и друго. ДМД е брзо прогресивна болест, а пациентите обично ја губат способноста за одење во текот на тинејџерските години помеѓу 6 и 13 години, а подоцна, во доцните тинејџерски до раните дваесетти години, имаат потреба од помош при дишење поради слабост на респираторните мускули.
Другите симптоми можат да вклучуваат:
кај лицата со ДМД можат да се развијат контрактури или вкочанети зглобови, сколиоза (искривување на рбетот) кои доведуваат до дополнително губење на подвижноста и функцијата
големи, месести мускули (псевдохипертрофија)
поради слабост на респираторните мускули, лицата со ДМД можат да имаат потешкотии со дишењето и да бидат изложени на зголемен ризик од респираторни инфекции
кардиомиопатија или слабост на срцевиот мускул е уште една честа компликација, па лицата со ДМД можат да доживеат проблеми со срцето, како што се аритмии или срцева слабост
Дијагнозата на ДМД обично се поставува врз основа на комбинација на медицинска историја, клинички преглед и други наодии. При дијагностицирање, најчесто се користат следните чекори:
физикален преглед за да се процени силата и функцијата на мускулите, одењето и присуството на контрактури
семејна историја на ДМД или други мускулни дистрофии може да сугерира присуство на болеста
тестови на крвта можат да се користат за мерење на нивото на креатин киназа, ензим кој се ослободува во крвотокот кога мускулното ткиво е оштетено. Зголемените нивоа на креатин киназа можат да укажуваат на оштетување на мускулите, што е вообичаен знак кај ДМД
мускулна биопсија, земање мал примерок на мускулно ткиво за анализа; таа може да покаже отсуство или намалено ниво на протеинот дистрофин, што е карактеристично за ДМД
генетско тестирање се користи за да се идентификуваат грешки (мутации, делеции, дупликации) во генот DMD
Поставувањето рана дијагноза на ДМД е неопходна за да се стопира прогресијата на болеста и да се подобри квалитетот на живот. Диференцијалната дијагноза вклучува Бекерова мускулна дистрофија и Лимб Гирдл (Limb Girdl) мускулна дистрофија.
ПОСЛЕДИЦИ
Појавата на првите симптоми и прогресијата на болеста можат да варираат од лице до лице и можат да напредуваат со различна брзина под влијание на голем број фактори, вклучувајќи ги специфичните генетски мутации и присуството на други медицински состојби. Без соодветен третман, просечниот животен век на лицата со ДМД е околу 25 години, а кардиомиопатијата и респираторната инсуфициенција се причини за рана смрт. Сепак, последните податоци од медицинските истражувања и напредокот во медицинската нега даваат надеж за подобрување на третманите, продолжување на животниот век и унапредување на квалитетот на живот.
ТРЕТМАН
Во моментов, не постои лек за целосно лекување на ДМД, но постојат третмани кои можат да помогнат во управувањето на симптомите и подобрување на квалитетот на живот. Третманот може да вклучува физикална терапија, ортопедски помошни уреди, а во некои случаи и лекови за подобрување на функцијата на мускулите и работата на срцето. За оваа болест, од суштинско значење е мултидисциплинарната нега.
Кортикостероидите се најчесто користените лекови кои помагаат да се стопира прогресијата на мускулната слабост и да се подобри мускулната функција. Физикалната терапија и рехабилитација се важни за одржување на подвижноста, флексибилноста и силата. Програмите за вежбање и помошните уреди можат да им помогнат на лицата со ДМД да ја задржат својата независност и подвижност што е можно подолго време. Помошни уреди, како што се инвалидски колички, скутери и рамки за стоење, можат да им помогнат на лицата со ДМД да ја задржат својата независност и подвижност. Неинвазивна вентилација и други форми на респираторна поддршка можат да помогнат во справување со проблемите со дишење. Срцевата поддршка често е потребна поради проблемите со работата на срцето.
Постојат неколку различни пристапи кои моментално се во фаза на истражување за лекување на ДМД:
Генска терапија е релативно нов пристап за лекување на ДМД кој има за цел да внесе функционална копија на генот за дистрофин во мускулните клетки, што може да помогне да се стопира прогресијата на болеста. Ова може да се направи со помош на вирусни вектори, модифицирани аденовируси, кои можат да го пренесат функционалниот ген во клетките. Во тек се клинички испитувања за да се процени безбедноста и ефикасноста на овој пристап
Други третмани во фаза на истражување имаат за цел да ги заобиколат мутациите во генот за дистрофин преку користење на антисенс олигонуклеотиди за отстранување на специфични егзони, градежните блокови на генот за дистрофин, од конечниот производ на mRNA. Ова може да резултира со производство на скратен, но функционален дистрофин
Терапијата со матични клетки има за цел да ги замени оштетените мускулни клетки со здрави, функционални мускулни клетки кои произведуваат дистрофин. Ова може да се направи преку користење на матични клетки, кои имаат способност да се развијат во различни типови на клетки, вклучувајќи ги и мускулните клетки
Важно е да се напомене дека специфичниот план за третман ќе зависи од сериозноста на симптомите и прогресијата на болеста. Редовното следење и прилагодување на планот за лекување треба да биде неопходно бидејќи болеста е прогресивна.
ПРЕДИЗВИЦИ
Моментално, најголемите предизвици кај ДМД вклучуваат:
Недостаток на ефективни третмани: во моментов не постои лек за целосно лекување на ДМД, а достапните третмани се однесуваат само на лекување на некои од симптомите кои не ја стопираат прогресијата на болеста
Поставување на дијагноза: симптомите на ДМД можат да бидат слични со оние на другите мускулни нарушувања, што ја отежнува дијагнозата
Пристап до нега: ДМД влијае на мал број лица, што им го отежнува на некои поединци пристапот до специјализирана нега
Менаџирање на симптомите: ДМД доведува до прогресивна мускулна слабост, а лицата со ДМД можат да доживеат низа симптоми, вклучувајќи потешкотии со подвижноста, дишењето и работата на срцето. Овие симптоми можат да бидат тешки за менаџирање, особено кога болеста е во напредната фаза
Трошоци за нега: трошоците за грижа на лицата со ДМД можат да бидат високи, поради потребата од постојан медицински третман, рехабилитација и помошни уреди
Истражување: и покрај напредокот во разбирањето на биологијата на ДМД, има уште многу да се научи за болеста и како ефикасно да се лекува. Потребни се дополнителни истражувања за да се пронајде лек и да се подобри животот на лицата со ДМД